Introduction
Malignant hyperthermia (MH) is a pharmacogenetic emergency that poses ongoing challenges for anesthesiologists, surgeons, and perioperative teams around the world.1,2 Despite improvements in anesthetic safety, the risk of malignant hyperthermia (MH) remains due to its unpredictable nature and potentially severe consequences. This review highlights the ten essential facts for effectively managing MH during the perioperative period.
1. Historical Insight: From Tragedy to Targeted Therapy
The first detailed account of Malignant Hyperthermia (MH) as a distinct clinical syndrome emerged in the early 1960s.2 The introduction of dantrolene in the 1970s, the only specific antidote for MH, significantly improved outcomes. With proper protocols and access to the drug, mortality rates decreased from as high as 80% to less than 5%.1,3–5
2. Epidemiology: A Rare Event with Global Implications
Malignant hyperthermia (MH) episodes are rare, with reported incidences ranging from 1 in 5,000 to 1 in 150,000 exposures to general anesthesia. These rates depend on factors such as population, genetic background, and the prevalence of triggering agents.1,4,6,7 Young males are affected more frequently than females.4,6 There are regional variations, and the disorder is more frequently reported in Europe, North America, and certain Asian populations.7,8
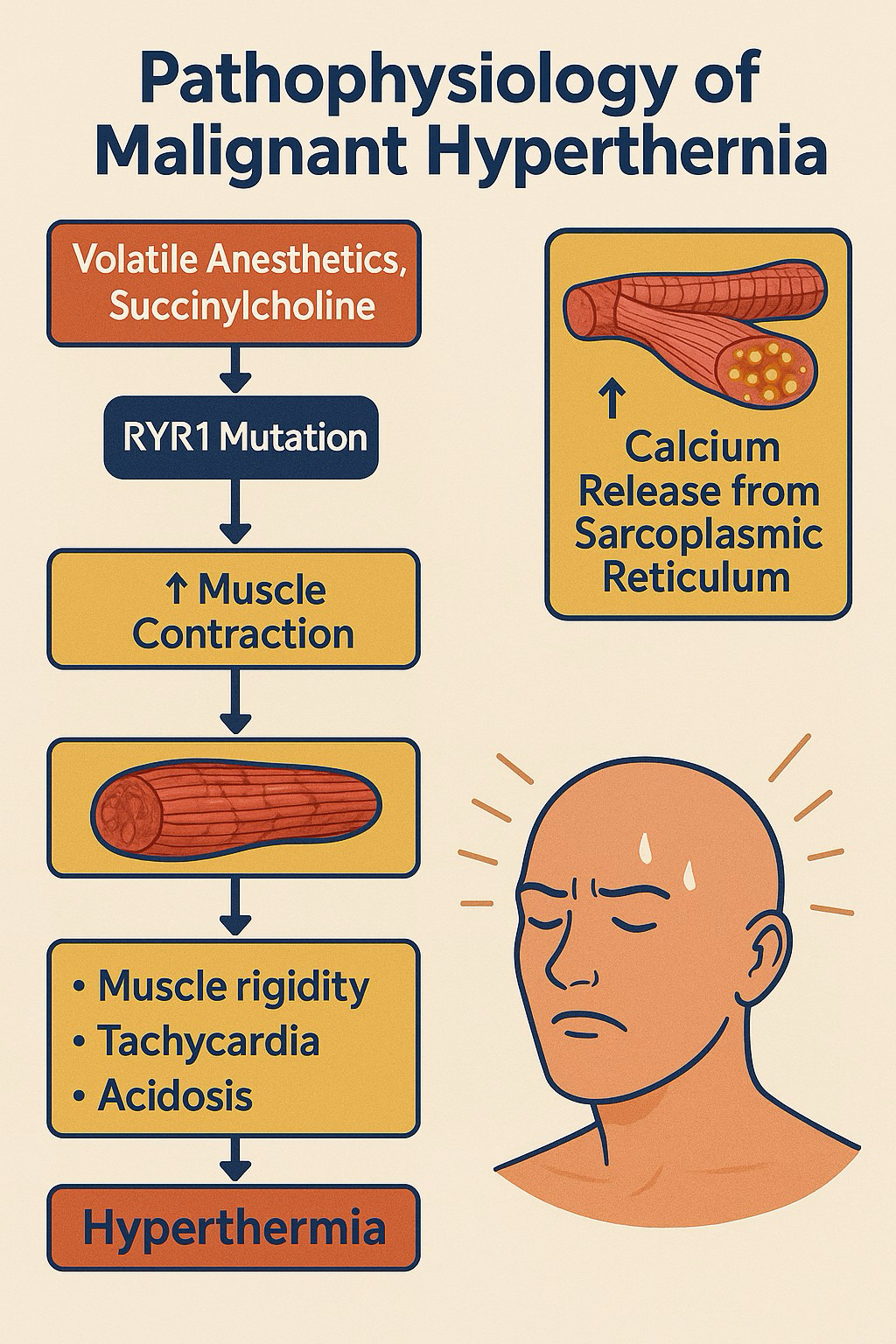
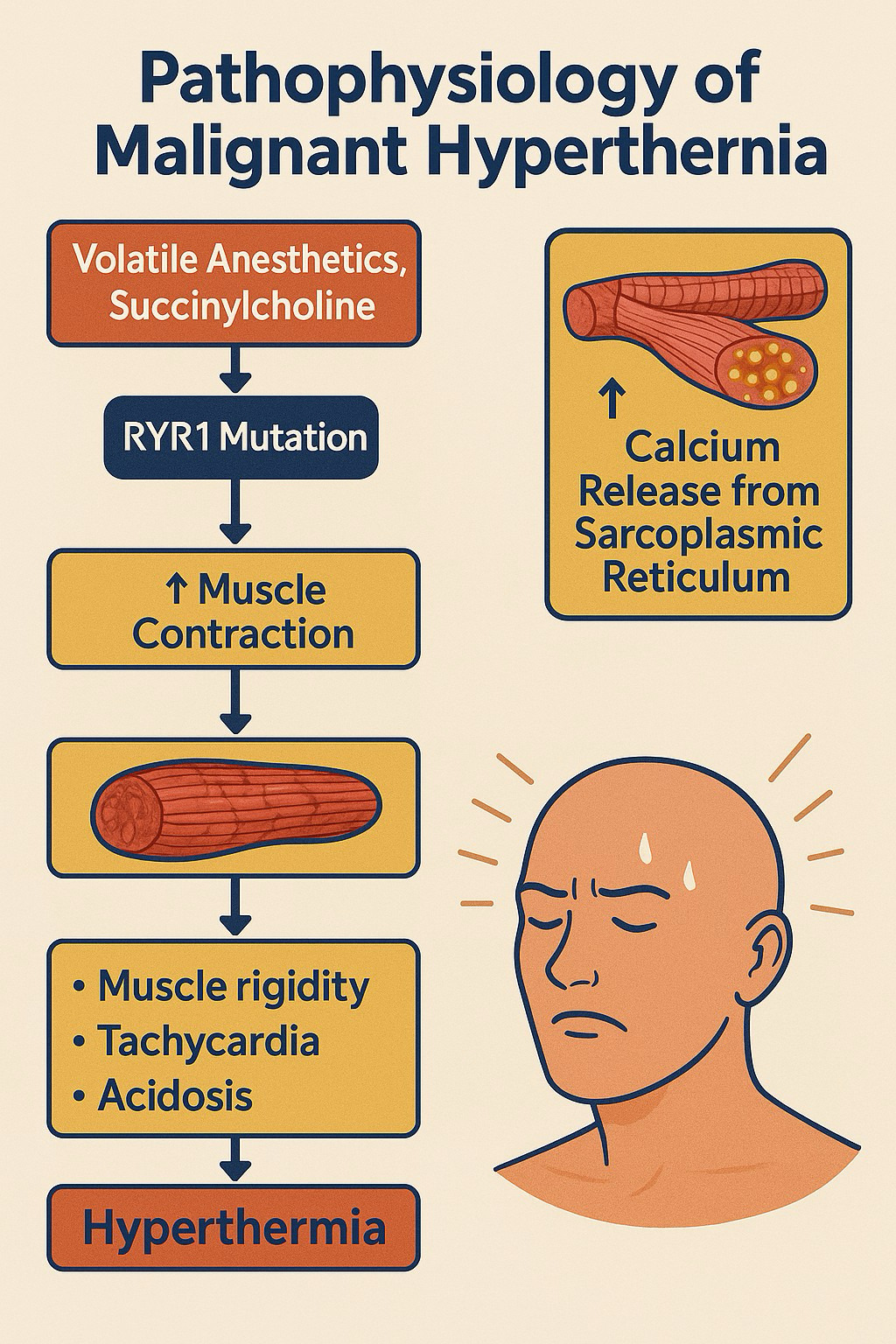
3. Pathophysiology: Unchecked Calcium—The Metabolic Engine of Crisis
MH is caused by a dysregulated intracellular calcium release in skeletal muscle, most commonly due to a defective ryanodine receptor (RyR1) in the sarcoplasmic reticulum.1–3,9,10 When exposed to triggering agents, the RyR1 channel remains open, leading to an influx of calcium ions into the myoplasm (Figure 1). This surge of calcium activates the muscle’s contractile machinery, significantly increasing metabolic rate, oxygen consumption, ATP hydrolysis, and heat production. If this process goes unchecked, it can result in lactic acidosis, rhabdomyolysis, hyperkalemia, and damage to multiple organs.1,2,9
4. Genetics: Dominant Inheritance and Heterogeneity
MH is primarily inherited in an autosomal dominant manner211]. The majority of human MH susceptibility is associated with mutations in the RYR1 gene on chromosome 19q13.1.1,10–12 Less commonly, mutations in CACNA1S (dihydropyridine receptor α1α1-subunit) and STAC3 play a role.11,12 Genetic testing exists but does not capture all cases of susceptibility, and negative genetic tests do not rule out MH risk.12,13
5. Triggering Agents: The Must-Know Provocateurs
MH crises are precipitated by specific anesthetic drugs—namely volatile inhalational agents (sevoflurane, desflurane, isoflurane, halothane) and the depolarizing muscle relaxant succinylcholine.1,4,11,14 Notably, non-depolarizing muscle relaxants, nitrous oxide, and intravenous anesthetics—including propofol, opioids, benzodiazepines, and ketamine—are safe in susceptible individuals, with extensive studies demonstrating propofol’s non-triggering effects on skeletal muscle calcium regulation1,214]. Agents such as succinylcholine can also trigger MH (up to 15% of cases).4
6. Clinical Presentation: Early Warning Signs and Catastrophic Progression
Acute MH episodes often begin intraoperatively but may rarely present postoperatively.4,15,16 The earliest hallmark is a sudden, unexpected rise in end-tidal carbon dioxide (ETCO2), out of proportion to ventilation.17 This condition may be accompanied by (Table 1):
-
Sinus tachycardia
-
Generalized muscle rigidity (notably masseter spasm after succinylcholine)
-
Hyperthermia (often a late sign, with temperature rising precipitously)
-
Metabolic and respiratory acidosis
-
Hyperkalemia
-
Myoglobinuria and rhabdomyolysis
-
Arrhythmias progressing to cardiac arrest in severe cases
Importantly, hyperthermia is a late sign; early diagnosis hinges on recognizing unexplained hypercapnia and muscle rigidity.4,6,16,17
7. Rapid Recognition, Differential Diagnosis, and Laboratory Clues
The diagnosis of MH is clinical, supported by laboratory findings of hyperkalemia, acidosis, elevated creatine kinase, and myoglobinuria.1,4,6,9 Differential diagnoses (Table 2) include conditions like Thyroid storm, Neuroleptic malignant syndrome, and more.
8. Immediate Crisis Management: Dantrolene and Supportive Measures
Time is crucial in MH. Immediate steps are1,2,4,9:
-
Stop all triggering agents.
-
Call for help and mobilize MH crisis protocol.
-
Administer dantrolene sodium IV: initial dose 2.5 mg/kg, repeated as needed, up to 10 mg/kg total if symptoms persist.
-
Hyperventilate with 100% oxygen (≥10 L/min) to address hypercapnia.
-
Actively cool the patient.
-
Treat hyperkalemia (with insulin/glucose, calcium, bicarbonate).
-
Manage dysrhythmias appropriately; avoid calcium channel blockers in conjunction with dantrolene due to the risk of hyperkalemia.
-
IV fluid therapy and vasopressors to support hemodynamics.
-
Insert a Foley catheter for urine output and detection of myoglobinuria.
-
Monitor Creatine Kinase, potassium, renal function, and coagulation parameters.
-
Prepare for transfer to ICU.
Dantrolene acts by inhibiting abnormal calcium release from the sarcoplasmic reticulum, directly reversing the underlying metabolic derangement. Guidelines demand its ready availability in all anesthetizing areas.1,2,4–6,9,18
9. Postoperative Care: Vigilance for Recurrence
After crisis control, intensive monitoring in an ICU is recommended for at least 24–48 hours, as up to 25% of patients may experience recrudescence.1,4,9 Dantrolene should be continued (1 mg/kg every 6 hours IV, or 0.25 mg/kg/hr infusion) as guided by symptoms. Monitoring for complications of rhabdomyolysis, hyperkalemia, hepatic dysfunction, renal dysfunction, and DIC (Disseminated Intravascular Coagulation) is essential.1,4,9 The caffeine-halothane contracture test (CHCT) remains the gold standard but is not universally available.1,12,13 It is available in only 4 centers in North America.
10. Anesthetic Strategies for MH-Susceptible Patients
The paramount principle is strict avoidance of triggers. For elective procedures in susceptible patients:
-
Use non-triggering anesthesia: total intravenous anesthesia.1,2,14
-
Avoid volatile anesthetics and succinylcholine.
-
Prepare anesthesia equipment by removing vaporizers, flushing circuits (≥20 minutes with high fresh gas flow), or using charcoal filters to absorb residual inhalational agents.1,2
-
Maintain a high index of suspicion for any unexplained tachycardia, hypercapnia, or rigidity.
-
Ensure immediate access to dantrolene and crisis protocols.1,9,14
-
Record clear documentation of susceptibility; advise medical alert bracelets and inform family for evaluation.1,4
With such comprehensive measures, elective surgery can proceed safely in most MH-susceptible patients.1,2,9,14
Conclusion
Malignant hyperthermia (MH) is a rare but critical emergency in the perioperative setting. Thanks to advancements in care, based on historical lessons, molecular understanding, Malignant Hyperthermia Association of the United States (MHAUS) development of protocols, and coordinated crisis response, the risk of death has significantly decreased. To achieve the best possible outcomes for patients at risk of this serious condition, it is essential to recognize the signs early, administer dantrolene without delay, provide strict supportive care, and maintain careful monitoring during postoperative recovery as well as in future anesthesia plans.